August 7, 2015
Looking Inside the Biotech Black Box (Part 14)
Market Exclusivity – Regulatory Data
Market exclusivity is a critical component of valuation. When patent exclusivity falls short of the expected 10 plus years, regulatory data exclusivity may provide some extra time before generics, biosimilars or other competitors can be approved.

- NDAs or New Drug Applications are filed for new prescription drug products and must include a full clinical data package, manufacturing information and labelling.
- ANDAs or Abbreviated New Drug Applications are filed for each new generic drug and must include the necessary clinical data package (human bioequivalence study(ies)), manufacturing and labelling – average time from submission to approval is about 30 months. With only a few exceptions, generic drug products are deemed to be interchangeable with each other and the reference brand product.
- 505 (b)(2) applications must include a full clinical data package, manufacturing information and labelling but do rely to some extent on published literature (information not paid for or owned by the applicant). This type of application may be used for many types of new drug products including new strengths, dosing regimens, formulations, routes of administration, prodrugs, active metabolites and combination products.

- NCE (new chemical entity) exclusivity – this exclusivity is available only if the active ingredient has not previously been approved by the FDA. No regulatory submission by another party may be made until 5 years after NDA approval. An ANDA submission may be made after 4 years only if it contains a Paragraph IV certification that it does not infringe the listed patents or that the listed patents are invalid. This type of submission automatically triggers a statutory delay of an additional 30 months, meaning that the FDA cannot approve the ANDA until the earlier of 7.5 years from NDA approval and a final court decision on the patent infringement claims.
- New clinical study exclusivity – the new clinical study must be sponsored by the applicant and provides the data which is essential to approval by the FDA. No ANDA may be approved by the FDA for a period of 3 years, although other clinical indications and dosage forms may be approved.
- Biologics exclusivity – biologics are essentially all drug products not deemed to be small molecule, traditional pharmaceuticals. Generic biologics are called biosimilars. The FDA cannot approve a biosimilar until 12 years after approval of the reference biologic product, although it can start the review process on a biosimilar 4 years after the approval.
- Orphan drug exclusivity – orphan drug designation is granted to a drug by the FDA if the medical condition affects fewer than 200,000 people in the U.S. The key benefit of this designation is a 7-year term of market exclusivity during which no product with the same active ingredient and clinical indication can be approved by the FDA.
- Pediatric exclusivity – if the NDA filer conducts an acceptable study in children, whether successful or not, a 6-month period of exclusivity begins on the date that either the patent exclusivity (Orange Book) or regulatory exclusivity would expire.
- Infectious disease exclusivity – in order to spur development of new drugs for serious or life-threatening infections, the FDA will add a 5-year period of exclusivity to the regulatory exclusivity of any qualifying antibacterial or antifungal drug product.
- First-to-file generic drug market exclusivity – the first applicant to file a substantially complete ANDA with a Paragraph IV certification will be granted a 180-day period of market exclusivity during which no other ANDAs for that specific drug product will be approved. The period of exclusivity is granted regardless of the outcome of any patent litigation resulting from the ANDA filing.
Citizen Petitions do not have any defined period of exclusivity but can potentially impact the periods of regulatory exclusivity. In a typical move, a brand company will file a Citizen Petition requesting that the FDA not grant approval of an ANDA unless it contains some very specific types of bioequivalence studies which would probably not have been conducted by the ANDA filer. The reason given by the brand company would be to protect the public from potentially unsafe and non-equivalent drugs whereas the real reason is protect their market exclusivity. The FDA has guidelines and timelines for its handling of Citizen Petitions.
The basic system for regulatory exclusivity in the E.U. is much simpler.
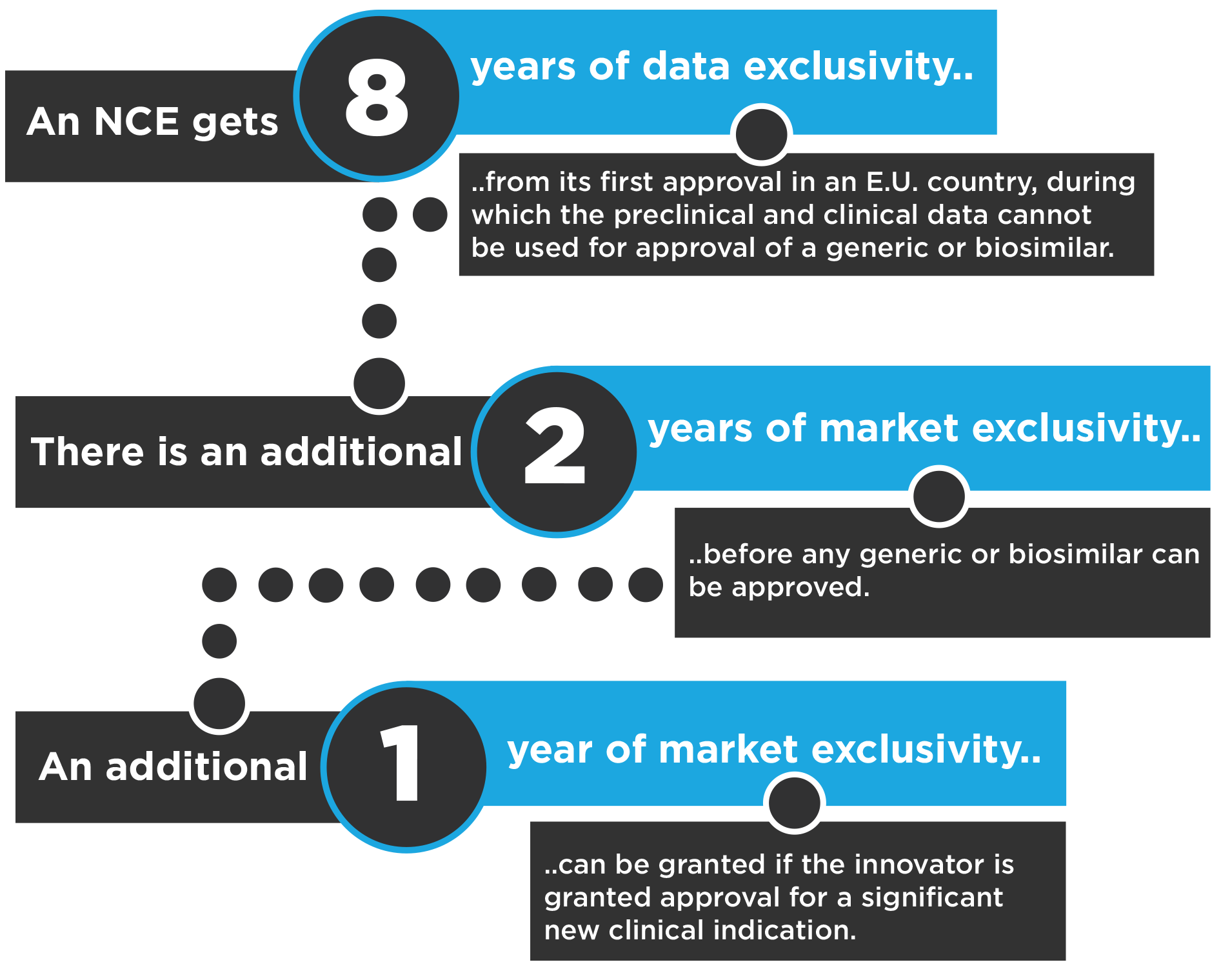
For example, an NCE approved initially for the treatment of pancreatic cancer and several years later for glioblastoma could have 11 years of combined data and market exclusivity in the E.U.
The regulatory exclusivity rules have impacted new drug development. There is an increased interest in biologics, triggered initially by the difficulty in making biosimilars and the lack of a U.S. regulatory pathway, then reinforced by the better U.S. regulatory exclusivity versus small molecules. There is also an increased interest in orphan drugs, probably a result of both the regulatory exclusivity and the extraordinary annual treatment costs, with several being around US$300,000.
In this and the prior blog, I have outlined the patent and regulatory rules which impact market exclusivity. In the next blog, some examples of product life-cycle management will be highlighted.
[The author and his immediate family members may have long or short positions in the shares of some companies mentioned in or assessed during the preparation of this blog. Past share price performance may not be an indicator of future share price performance. This blog does not consider the investment objectives, financial situation or particular needs of any particular person. Investors should obtain professional advice based on their own individual circumstances before making an investment decision.]
As with all our posts, please see our full legal disclaimer.


